






Medizinprodukte sind sowohl im gewerblichen als auch im privaten Bereich unersetzlich. Professionelle Anwendung finden sie beispielsweise in Krankenhäusern, ärztlichen Praxen und Rehakliniken. Auch in der ambulanten Pflege, in Altenpflegeheimen, Wohnheimen für Menschen mit Behinderung und integrativen Kindertagesstätten kommen Medizinprodukte zum Einsatz. Im Privatbereich haben beispielsweise Schlafapnoe-Geräte bei Schlaf- und nächtlicher Atmungsstörung oder „Medical Apps“ deutlichen Einzug gehalten.
Die Akteure: Für wen sind die Änderungen wichtig?
Ist die Umsetzung des vorher bereits bestehenden Regelwerkes noch unvollständig oder unzureichend im Betrieb erfolgt, so besteht jetzt erhöhter Handlungsbedarf, Informations- und Qualifikationsbedarf der verantwortlichen Personen. Im Fokus stehen diejenigen, die einen sicheren Umgang mit Medizinprodukten zum Schutz von Patienten, Beschäftigten oder Dritten gewährleisten müssen, also Betreiber und Anwender von Medizinprodukten. Im Folgenden wird die geänderte Rechtslage erläutert und zugleich auf bestehende, bleibende Pflichten hingewiesen.
Die Definition der Akteure ist im § 2 der Medizinprodukte-Betreiberverordnung (MPBetreibV) zu finden:
- „Gesundheitseinrichtung (…) ist jede Einrichtung, Stelle oder Institution, (…), in der Medizinprodukte durch medizinisches Personal, Personen der Pflegeberufe oder sonstige dazu befugte Personen berufsmäßig betrieben oder angewendet werden.“
- „Betreiber (…) ist jede (…) Person, die für den Betrieb der Gesundheitseinrichtung verantwortlich ist, in der das Medizinprodukt durch dessen Beschäftigte betrieben oder angewendet wird. (…) wer außerhalb von Gesundheitseinrichtungen in seinem Betrieb oder seiner Einrichtung oder im öffentlichen Raum Medizinprodukte zur Anwendung bereithält.“
- „Anwender ist, wer ein Medizinprodukt (…) am Patienten einsetzt.“
Herausforderungen für Betreiber und Anwender
Betreiber von Medizinprodukten, wie die Leitung einer Gesundheitseinrichtung, stehen Pflichten aus einem umfassenden Regelwerk gegenüber. Aktuell gibt es auf europäischer und nationaler Ebene Veränderungen in den Regelwerken für Medizinprodukte. Daraus ergeben sich für Betreiber von Gesundheitseinrichtungen – aber auch für beratende Fachkräfte für Arbeitssicherheit und Betriebsärzte – neue Herausforderungen. Die relevanten Regelwerke sollen hier vorgestellt und näher erörtert werden. Erläutert werden die Vorschriften, die für Betreiber und Anwender relevant sind.
Insgesamt betrachtet ist der Umgang mit den Regelwerken anspruchsvoller geworden:
- Die einzelnen Regelwerke haben eine Vielzahl von Adressaten wie Hersteller, Behörden usw. – was den Überblick der eigenen Pflichten, zum Beispiel als Betreiber, erschwert.
- Die bisherige Rechtslage erlaubte den Betreibern sich auf wenige nationale Regelwerke wie MPG, MPBetreibV und MPSV zu beschränken. Die zukünftigen Regelwerke sind sowohl auf europäischer Ebene als auch nationaler Ebene zu finden.
- Diese neuen Regelwerke sind nicht in sich selbsterklärend und abgeschlossen. Für die Zukunft wird für die Betreiber und Anwender mehr denn je die gleichzeitige Nutzung der für sie relevanten Regelwerke erforderlich sein.
- Die Regelwerke sind in ihren Namen, aber nicht in ihrem Inhalt zum Verwechseln ähnlich, wie zum Beispiel „Medizinprodukte-EU-Anpassungsgesetz“ und „Medizinprodukte-EU-Anpas-sungsverordnung“.
- Die Gesetzes- und Verordnungsnamen lassen oftmals nicht auf den Regelungsgegenstand oder den Adressaten schließen. So regelt die „Medizinprodukte-Anwendermelde- und Informationsverordnung“ vereinfacht gesagt den Umgang mit gefährlich gewordenen Medizinprodukten.
Die Regelwerke im Überblick – was sich konkret ändert
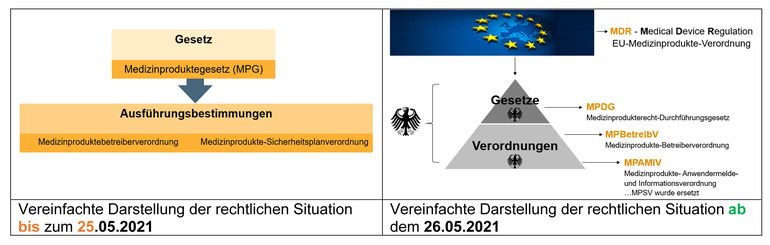
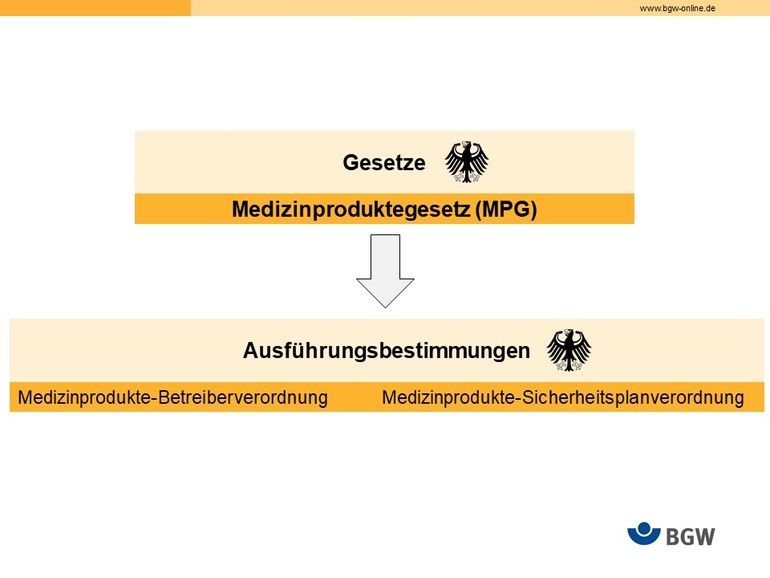
Bisher standen folgendes Gesetz und seine Ausführungsbestimmungen für Betreiber und Anwender im Vordergrund:
- MPG, Medizinproduktegesetz (entfällt)
- MPBetreibV, Medizinprodukte-Betreiberverordnung (gilt weiterhin)
- MPSV, Medizinprodukte-Sicherheitsplanverordnung (entfällt)
Unbestritten war der Einfluss von europäischen Vorgaben, die in die nationalen Regelwerke über die Jahre eingeflossen sind. Trotzdem war ein konzentrierter Blick auf diese drei nationalen Regelwerke bisher ausreichend, um die Betreiber- und Anwenderpflichten im Blick zu behalten.
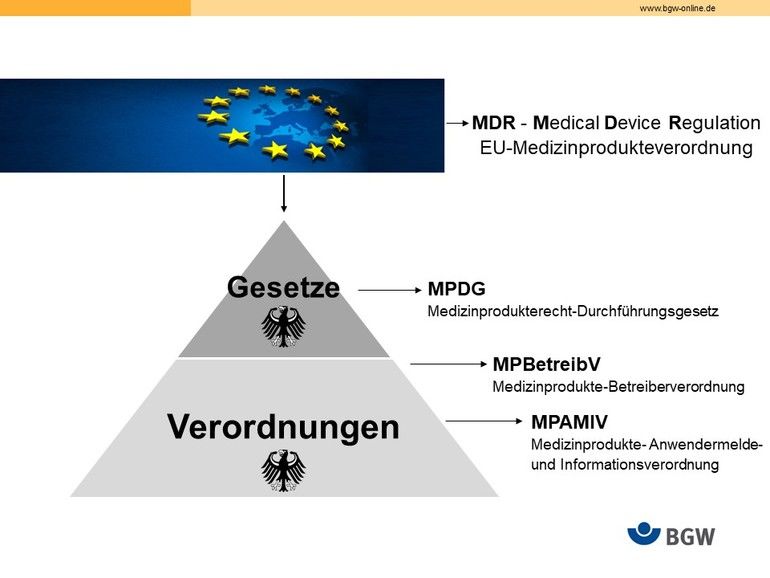
Seit dem 26.05.2021 sind folgende Regelwerke zu berücksichtigen:
- MDR, Europäische Medizinprodukteverordnung (NEU!)
- MPDG, Medizinprodukterecht-Durchführungsgesetz (NEU!)
- MPBetreibV, Medizinprodukte-Betreiberverordnung
- MPAMIV, Medizinprodukte-Anwendermelde- und Informationsverordnung (NEU!)
Die Regelwerke im Detail
Im folgenden Abschnitt sollen die einzelnen Regelwerke vorgestellt und die relevanten Inhalte für Betreiber und Anwender ausgearbeitet werden.
1. MDR – Die Europäische Medizinprodukteverordnung
Die MDR ist tragendes Fundament des Medizinprodukterechtes und löst gewissermaßen – mit ihrem Geltungsbeginn zum 26.05.2021 – eine Art Dominoeffekt aus. Die MDR setzt das bekannte nationale Medizinproduktgesetz in Teilen außer Kraft und löst einen hohen Anpassungsbedarf in vielen nationalen Regelwerken aus. Dieser Anpassungsbedarf hat eine Auswirkung auf die relevanten Regelwerke für Anwender und Betreiber.
Im Mittelpunkt der MDR stehen die Aspekte der Produktsicherheit, hohe Standards für Qualität und Sicherheit der Medizinprodukte“ und der „reibungslos funktionierende Binnenmarkt für Medizinprodukte“.
Für den Betreiber und Anwender dient die MDR vorrangig dem besseren Verständnis und klärt die grundlegenden Begriffe wie „Medizinprodukt“. Dabei formuliert die MDR keine unmittelbaren Forderungen und Pflichten an Anwender und Betreiber. Sie ist der gültige Rechtsbezug für diese Personengruppe und der Bezugspunkt der nationalen Gesetze und Verordnungen für Pflichten von Betreiber und Anwender.
2. MPDG – Medizinprodukterecht-Durchführungsgesetz
Das neue MPDG ergänzt die MDR und ist, vereinfacht dargestellt, der inhaltliche Nachfolger des bekannten Medizinproduktegesetzes (MPG). Das MPDG stellt ein Bindeglied zwischen der neuen MDR und den nationalen Gesetzen und Verordnungen untereinander dar.
Zu den wichtigsten Aspekten für Betreiber und Anwender gehören zweifellos die Paragraphen § 11 und § 12 des MPDG. Ohne die Begrifflichkeit zu verwenden, wird darin ein „Gefährdungsverbot“ ausgesprochen.
In § 11 MPDG heißt es: „Medizinprodukte dürfen nicht betrieben oder angewendet werden, wenn sie Mängel aufweisen, durch die Patienten, Beschäftigte oder Dritte gefährdet werden können.“
Der § 12 definiert den Begriff „Mangel“. Ein Mangel am Medizinprodukt liegt demnach bereits vor, wenn „das Datum abgelaufen ist, bis zu dem das Produkt sicher verwendet werden kann.“ An der Stelle wird das vorgegebene hohe Schutzniveau des Gesetzgebers deutlich – mit weitreichenden Konsequenzen! Denn im § 92 „Strafvorschrift“ heißt es: „Mit Freiheitsstrafe bis zu drei Jahren oder mit Geldstrafe wird bestraft, wer Medizinprodukte mit Mängel (…) betreibt oder anwendet, (…). Mit Freiheitsstrafe von (…) bis zu zehn Jahren wird bestraft, wer (…) die Gesundheit einer großen Zahl von Menschen gefährdet, einen anderen in die Gefahr des Todes oder einer schweren Schädigung an Körper oder Gesundheit bringt.“
Der § „94 Bußgeldvorschriften“ hält, wie es der Name vermuten lässt, ein umfangreiches Bußgeld bei Verstößen bereit. „Die Ordnungswidrigkeit kann mit einer Geldbuße bis zu dreißigtausend Euro geahndet werden.“ Ordnungswidrig handelt, wer vorsätzlich oder fahrlässig mangelhafte Medizinprodukte „in den Verkehr bringt, in Betrieb nimmt, betreibt oder anwendet“.
Hervorzuheben ist, dass nicht nur der Betreiber, sondern auch der Anwender von „mangelhaften“ Medizinprodukten Adressat des Gefährdungsverbotes und von Sanktionsmaßnahmen nach MPDG ist. In der betrieblichen Praxis sehen sich klassische Anwender, wie zum Beispiel ambulante Pflegedienste, die Medizinprodukte des Patienten beziehungsweise des zu Pflegenden (Pflegebett, Blutzuckermessgerät, Lifter) benutzen, oftmals in keiner Verpflichtung, da sie das Medizinprodukt „nur verwenden“ und „das Medizinprodukt nicht ihnen gehört“. Dieser Umstand muss angesichts der bedrohlich wirkenden Sanktionsmöglichkeiten für Anwender neu überdacht werden.
Insbesondere im Zusammenhang des §11 MPDG wird es für Beschäftigte als Anwender brenzlig, wenn diese von Patienten oder Dritten mitgebrachte, aber ungeprüfte (z. B. fehlende sicherheitstechnische Kontrollen nach § 11 MPBetreibV) Medizinprodukte anwenden, in deren Gebrauch sie zudem nicht mal (nach §4; §10 MPBetreibV) eingewiesen worden sind. Hierbei kann es sich um mitgebrachte
Medizinprodukte eines Patienten in einer stationären Einrichtung (Altenpflege, Krankenhaus) handeln oder um vorgefundene Medizinprodukte in der Häuslichkeit des zu Pflegenden in der ambulanten Pflege, in beiden Fällen wäre als Beispiel das private Blutzuckermessgerät des Patienten zu nennen.
3. MPBetreibV – Medizinprodukte-Betreiberverordnung
Was für Arbeitsmittel die Betriebssicherheitsverordnung ist, ist für Medizinprodukte die Medizinprodukte-Betreiberverordnung, kurz MPBetreibV. Sie regelt nahezu alle Verpflichtungen des Betreibers und Anwenders für den sicheren Umgang mit Medizinprodukten und stellt für die betriebliche Praxis das wichtigste Instrument und Regelwerk dar. Dreh- und Angelpunkt der Verordnung ist, ob man sich im Adressatenkreis des „Betreibers“, „Anwenders“ oder der „Gesundheitseinrichtung“ nach der Definition der MPBetreibV wiederfindet, da nahezu jeder Paragraph der MPBetreibV sich an einen dieser Adressaten richtet. Eine kurze Übersicht der Pflichten (nicht abschließend) nach der MPBetreibV:
- Einweisung von Medizinprodukten (§ 4 Abs. 3 MPBetreibV)
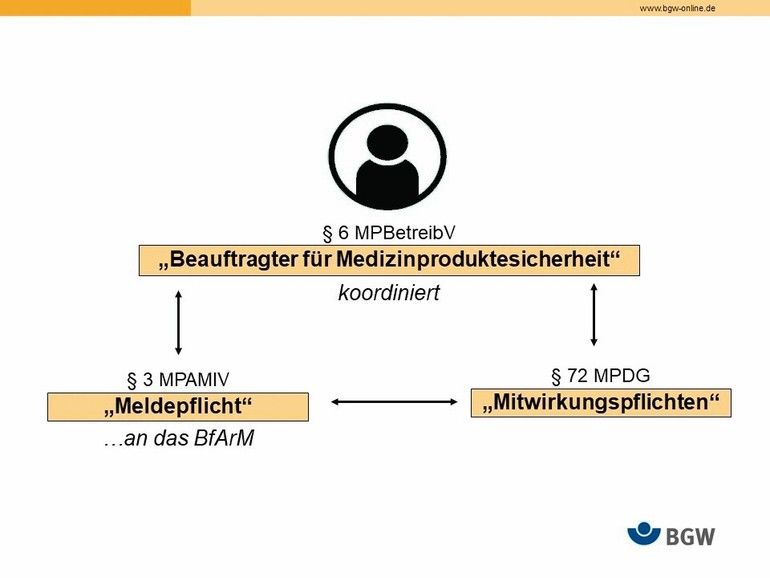
- Beauftragter für Medizinproduktesicherheit (§ 6 MPBetreibV)
- Besondere Einweisung von Medizinprodukten (§ 10 MPBetreibV)
- Prüfung von Medizinprodukten (sicherheitstechnische Kontrollen nach § 11 MPBetreibV, messtechnische Kontrollen nach § 14 MPBetreibV)
- Bestandsverzeichnis (§ 13 MPBetreibV)
- Medizinproduktebuch (§ 12 MPBetreibV)
Führungskräfte von Einrichtungen und Träger des Gesundheitsdienstes, die die umfassende Novellierung der MPBetreibV zum 01.01.2017 wahrgenommen und ihre Organisation im Hinblick auf die Pflichten angepasst haben, werden durch die MDR und die neuen Regelwerke nur geringfügige Veränderungen wahrnehmen. Zu den bereits seit 2017 hinzugekommenen Pflichten gehören der „Beauftragte für Medizinproduktesicherheit“ nach § 6 MPBetreibV sowie deren geforderte Veröffentlichung der E‑Mail-Adresse auf der Unternehmens-homepage.
Die meisten Veränderungen betreffen die Anknüpfung der MPBetreibV an die MDR, MPDG und die neue MPAMIV. Insgesamt ergeben sich rein aus der MPBetreibV keine neuen Pflichten, noch der Wegfall der bisher bestehenden Pflichten.
Die für die Zukunft zu erwartenden Änderungen betreffen die MPBetreibV selbst. Zukünftig wird der Rückverfolgbarkeit von Medizinprodukten anhand eines Systems der einmaligen Produktkennung „UDI-System“ (Unique Device Identification System) auf dem Produkt selbst oder seiner Verpackung eine große Bedeutung zukommen.
Naheliegend wird dieses UDI-System in das vom Betreiber geführte Bestandsverzeichnis oder bei der Meldepflicht von „mangelhaften“ Medizinprodukten Einzug halten.
Für die Zukunft sind somit Entwicklungen absehbar, die eine Anpassung der MPBetreibV notwendig machen.
4. MPAMIV – Medizinprodukte-Anwendermelde- und Informations-verordnung
Die MPAMIV regelt unter anderem die Meldung von „mutmaßlichen schwerwiegenden Vorkommnissen“ neu und ersetzt die bisherige Medizinprodukte-Sicherheitsplanverordnung, kurz MPSV, vollständig.
Dabei ist unter einem „mutmaßlichen schwerwiegenden Vorkommnis“ vereinfacht eine betriebliche Situation im Zusammenhang mit einem Medizinprodukt zu verstehen, bei dem Patienten, Beschäftigte oder Dritte zu Schaden hätten kommen können, dies schließt „Anwendungsfehler aufgrund ergonomischer Merkmale oder einer Unzulänglichkeit der vom Hersteller bereitgestellten Informationen“ mit ein.
Im Hinblick auf die „mutmaßlichen schwerwiegenden Vorkommnisse“ lässt sich die Komplexität und Verzahnung des gesamten Regelwerkes im Medizinprodukterecht aufzeigen (siehe Abb. 2). Während die MPAMIV in § 3 die Meldepflichten von „mutmaßlichen schwerwiegenden Vorkommnissen“ gegenüber Betreibern und Anwendern fordert, regelt das MPDG im § 72 eine „Mitwirkungspflicht, sodass Medizinprodukte, „die im Verdacht stehen, an einem schwerwiegenden Vorkommnis beteiligt zu sein, nicht verworfen werden, bis die Risikobewertung der zuständigen Bundesoberbehörde abgeschlossen ist“.
Die Koordination dieser oben genannten innerbetrieblichen Prozesse und Pflichten wird wiederum von „Beauftragten für Medizinproduktesicherheit“ nach § 6 MPBetreibV (nicht gleichzusetzen mit den Medizinproduktebeauftragten) realisiert.
Die nun vorliegende Besonderheit und gleichzeitige Schwierigkeit ist, dass Betreiber und Anwender gewohnt waren, die Pflichten im Umgang mit „mutmaßlichen schwerwiegenden Vorkommnissen“ einzig in der MPSV vorzufinden. Nach dem Ungültigwerden der MPSV erstrecken sich nun fast identische Inhalte auf die neue MPAMIV und das MPDG.
Fazit
Welche nächsten Schritte aufgrund der Änderungen durch die MDR im Betrieb notwendig und sinnvoll sind, ist vom bisherigen Umsetzungstand der Regelwerke in die betriebliche Praxis abhängig. Ist die MPBetreibV nach ihrer Novellierung in 2017 im Betrieb umgesetzt, finden sich nahezu identische Pflichten in neuen Regelwerken. Insgesamt betrachtet sind inhaltliche Veränderungen geringen Umfangs für Betreiber und Anwender wahrzunehmen. Der zeitliche Anpassungsaufwand für Betriebe ist hierbei überschaubar, auch wenn die Komplexität des Regelwerkes für die Zukunft anspruchsvoller geworden ist.
Ist die Umsetzung des vorher bereits bestehenden Regelwerkes noch unvollständig oder unzureichend im Betrieb erfolgt, so besteht jetzt erhöhter Handlungsbedarf, Informations- und Qualifikationsbedarf der verantwortlichen Personen. Sonst schieben Einrichtungen eine große „Bugwelle“ aus alten und neuen Betreiber- und Anwenderpflichten vor sich her und der Durchblick fällt zunehmend schwer.
Fachkräfte für Arbeitssicherheit und Betriebsärzte, welche die Branchen des Gesundheitswesens wie Arztpraxen, Krankenhäuser, Altenpflegeheime nach Arbeitssicherheitsgesetz und DGUV Vorschrift 2 betreuen und beraten, könnten den Unternehmer im Arbeitsschutzausschuss zu den Neuerungen im Medizinprodukterecht informieren, die Umsetzung im eigenen Betrieb mit medizinischen Fachkräften überprüfen und Handlungsbedarf ableiten.
Ferner wäre eine Weiterqualifikation von Personen mit der Verantwortung der Betreiberpflichten für Medizinprodukte denkbar. Diese bietet beispielsweise das Seminar „Medizinprodukte sicher betreiben und anwenden“ der Berufsgenossenschaft für Gesundheitsdienst und Wohlfahrtspflege (BGW). Mehr dazu auf www.bgw-online.de mit dem Seminarkürzel „W10“ im Suchfeld.
Für die Zukunft bleibt es spannend, welche weiteren Veränderungen durch die MDR Auswirkung auf die relevanten Regelwerke für Betreiber und Anwender haben.
 Foto: privat
Foto: privatAutor: Dipl.-Ing. Michael Kowatzky
Aufsichtsperson bei der Berufsgenossenschaft für Gesundheitsdienst und Wohlfahrtspflege (BGW)
E‑Mail: michael.kowatzky@bgw-online.de








{kind=link}